
ALECENSA 150 mg, gélule, boîte de 28 plaquettes thermoformées de 8
Dernière révision : 17/10/2024
Taux de TVA : 2.1%
Prix de vente : 3 836,60 €
Taux remboursement SS : 100%
Base remboursement SS : 3 836,60 €
Laboratoire exploitant : ROCHE
Source :

Traitement adjuvant du cancer bronchique non à petites cellules (CBNPC) réséqué
Alecensa est indiqué en monothérapie dans le traitement adjuvant, après résection complète de la tumeur, des patients adultes atteints d'un cancer bronchique non à petites cellules (CBNPC) ALK- positif à haut risque de récidive (voir rubrique Propriétés pharmacodynamiques pour les critères de sélection).
Traitement du CBNPC avancé
Alecensa est indiqué en monothérapie en première ligne de traitement des patients adultes atteints d'un CBNPC avancé ALK-positif.
Alecensa est indiqué en monothérapie dans le traitement du CBNPC avancé ALK-positif chez les patients adultes préalablement traités par crizotinib.
Hypersensibilité à l'alectinib ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Pneumopathie interstitielle diffuse / Pneumopathie inflammatoire
Des cas de pneumopathie interstitielle diffuse / pneumopathie inflammatoire ont été rapportés lors des essais cliniques avec Alecensa (voir rubrique Effets indésirables). Les patients doivent faire l'objet d'une surveillance afin de déceler tout symptôme pulmonaire évocateur d'une pneumopathie. Alecensa doit être immédiatement interrompu en cas de diagnostic de pneumopathie interstitielle diffuse / pneumopathie inflammatoire et doit être définitivement arrêté si aucune autre cause potentielle de pneumopathie interstitielle diffuse / pneumopathie inflammatoire n'a été identifiée (voir rubrique Posologie et mode d'administration).
Hépatotoxicité
Des augmentations des taux d'alanine aminotransférase (ALAT) et d'aspartate aminotransférase (ASAT) supérieures à 5 fois la limite supérieure de la normale (LSN), ainsi que des augmentations du taux de bilirubine supérieures à 3 fois la LSN sont survenues chez des patients inclus dans des essais cliniques pivots avec Alecensa (voir rubrique Effets indésirables). La majorité de ces événements est survenue pendant les 3 premiers mois de traitement. Dans les essais cliniques pivots d'Alecensa, il a été rapporté que trois patients avec une augmentation des ASAT ou ALAT de Grade 3-4 avaient des lésions hépatiques d'origine médicamenteuse. Des augmentations simultanées des ALAT et ASAT supérieures ou égales à 3 fois la LSN et de la bilirubine totale supérieure ou égale à 2 fois la LSN, avec des phosphatases alcalines normales, ont été rapportées chez un patient traité par Alecensa dans les essais cliniques.
La fonction hépatique, comprenant un dosage des ALAT, ASAT et de la bilirubine totale, doit faire l'objet d'une surveillance à l'initiation du traitement puis toutes les 2 semaines pendant les 3 premiers mois de traitement. Par la suite, la surveillance doit être effectuée périodiquement, puisque des événements peuvent survenir au-delà de 3 mois de traitement, avec un test plus fréquent chez les patients qui présentent une augmentation des aminotransférases et de la bilirubine. En fonction de la sévérité des effets indésirables, le traitement par Alecensa doit être interrompu puis repris au palier de dose inférieur, ou arrêté définitivement tel que décrit dans le Tableau 2 (voir rubrique Posologie et mode d'administration).
Myalgie sévère et augmentation des créatines phosphokinases (CPK)
Des myalgies ou des douleurs musculo-squelettiques ont été rapportées chez des patients dans les essais pivots avec Alecensa, y compris des événements de Grade 3 (voir rubrique Effets indésirables).
Une augmentation des CPK a été rapportée dans les essais pivots avec Alecensa, y compris des événements de Grade 3 (voir rubrique Effets indésirables). Le délai médian de survenue des augmentations des CPK de Grade ≥ 3 était de 15 jours dans les essais cliniques (BO40336, BO28984, NP28761, NP28673).
Les patients doivent être informés qu'ils doivent déclarer toute douleur, sensibilité ou faiblesse musculaire inexpliquées. Le taux des CPK doit être évalué toutes les deux semaines durant le premier mois de traitement et lorsqu'il est cliniquement indiqué chez des patients déclarant des symptômes.
Selon la sévérité de l'augmentation des CPK, le traitement par Alecensa doit être interrompu, puis repris à la même dose ou à une dose inférieure (voir rubrique Posologie et mode d'administration).
Bradycardie
Des cas de bradycardie symptomatique peuvent survenir avec Alecensa (voir rubrique Effets indésirables). La fréquence cardiaque et la pression artérielle doivent être surveillées si cliniquement indiqué. Une adaptation de la posologie n'est pas nécessaire en cas de bradycardie asymptomatique (voir rubrique Posologie et mode d'administration). Si les patients présentent une bradycardie symptomatique ou des événements menaçant le pronostic vital, l'utilisation de traitements concomitants favorisant la bradycardie et de traitements antihypertenseurs doit être évaluée et le traitement par Alecensa doit être adapté tel que décrit dans le Tableau 2 (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions, « Les substrats de la glycoprotéine P (P-gp) » et « Les substrats de la protéine de résistance du cancer du sein (BCRP) »).
Anémie hémolytique
Des cas d'anémie hémolytique ont été rapportés avec Alecensa (voir rubrique Effets indésirables). Si la concentration d'hémoglobine est inférieure à 10 g/dL et qu'une anémie hémolytique est suspectée, le traitement par Alecensa doit être interrompu et des analyses de laboratoire appropriées doivent être initiées. Si l'anémie hémolytique est confirmée, le traitement par Alecensa doit être repris au palier de dose inférieur dès la résolution tel que décrit dans le Tableau 2 (voir rubrique Posologie et mode d'administration).
Perforation gastrointestinale
Des cas de perforations gastrointestinales ont été rapportés chez des patients à risque (par ex : antécédent de diverticulite, métastases du tractus gastrointestinal, utilisation concomitante de médicaments présentant un risque connu de perforation gastrointestinale) traités par alectinib. L'interruption du traitement par Alecensa doit être envisagée chez les patients présentant une perforation gastrointestinale. Les patients doivent être informés des signes et symptômes de perforations gastrointestinales et il doit leur être conseillé de consulter rapidement en cas de survenue de tels signes et symptômes.
Photosensibilité
Des cas de photosensibilité à la lumière du jour ont été rapportés après une administration d'Alecensa (voir rubrique Effets indésirables). Il faut conseiller aux patients d'éviter l'exposition prolongée au soleil pendant le traitement par Alecensa, et jusqu'à au moins 7 jours après l'arrêt du traitement. Il faut également conseiller aux patients d'appliquer un écran solaire à large spectre anti-Ultraviolet A (UVA)/Ultraviolet B (UVB) et un baume pour les lèvres (Indice de protection solaire [SPF] ≥ 50) afin de se protéger contre les érythèmes solaires.
Toxicité embryo-fœtale
Alecensa peut entraîner des malformations fœtales lorsqu'il est administré à une femme enceinte. Les patientes en âge de procréer recevant Alecensa doivent utiliser des méthodes contraceptives hautement efficaces durant le traitement et pendant au moins 5 semaines après l'arrêt du traitement par Alecensa (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions, Fertilité, grossesse et allaitement et Données de sécurité préclinique). Les patients de sexe masculin ayant des partenaires en âge de procréer doivent utiliser des méthodes contraceptives hautement efficaces durant le traitement et pendant au moins 3 mois après l'arrêt du traitement par Alecensa (voir rubriques Fertilité, grossesse et allaitement et Données de sécurité préclinique).
Intolérance au lactose
Ce médicament contient du lactose. Son utilisation est déconseillée chez les patients présentant une intolérance au galactose, un déficit en lactase de lapp ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares).
Contenu en sodium
Ce médicament contient 48 mg de sodium par dose journalière (1200 mg), ce qui équivaut à 2,4 % de la dose journalière maximale de 2 g recommandée par l'OMS pour un adulte.
Résumé du profil de tolérance
Les données présentées ci-dessous reflètent l'exposition à Alecensa chez 533 patients atteints d'un CBNPC réséqué ou avancé ALK-positif. Ces patients ont reçu Alecensa à la dose recommandée de 600 mg deux fois par jour dans les essais cliniques pivots pour le traitement adjuvant du CBNPC réséqué (BO40336, ALINA) ou du CBNPC avancé (BO28984, ALEX; NP28761; NP28673). Voir rubrique Propriétés pharmacodynamiques pour plus d'informations sur les participants des essais cliniques.
Dans l'essai BO40336 (ALINA ; N =1 28), la durée médiane d'exposition à Alecensa était de 23,9 mois. Dans l'essai BO28984 (ALEX ; N = 152), la durée médiane d'exposition à Alecensa était de 28,1 mois. Dans les essais cliniques de phase II (NP28761, NP28673 ; N = 253), la durée médiane d'exposition à Alecensa était de 11,2 mois.
Les effets indésirables les plus fréquents (≥ 20 %) étaient : constipation, myalgie, œdème, anémie, éruption cutanée, augmentation de la bilirubinémie et augmentation des ALAT et augmentation des ASAT.
Tableau des effets indésirables
Le tableau 3 liste les effets indésirables survenus chez les patients ayant reçu Alecensa au cours des essais cliniques (BO40336, BO28984, NP28761, NP28673).
Les effets indésirables listés dans le Tableau 3 sont présentés par classe de système d'organes et par catégorie de fréquence définie selon les conventions suivantes : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000). Dans chaque classe de système d'organes, les effets indésirables sont présentés par ordre décroissant de fréquence et de sévérité. Au sein du même groupe de fréquence et de sévérité, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 Effets indésirables signalés chez les patients traités par Alecensa au cours d'essais cliniques (BO40336, BO28984, NP28761, NP28673 ; N=533).
|
Classe de système d'organes |
Alecensa |
|
|
Catégorie de fréquences (tout grade) |
Catégorie de fréquences (grades 3-4) |
|
|
Affections hématologiques et du système lymphatique |
||
|
Anémie1) |
Très fréquent |
Fréquent |
|
Anémie hémolytique2) |
Fréquent |
-* |
|
Affections du système nerveux |
||
|
Dysgueusie3) |
Fréquent |
Peu fréquent |
|
Affections oculaires |
||
|
Trouble de la vision4) |
Fréquent |
-* |
|
Affections cardiaques |
||
|
Bradycardie5) |
Très fréquent |
-* |
|
Affections respiratoires, thoraciques et médiastinales |
||
|
Pneumopathie interstitielle diffuse / pneumopathie inflammatoire |
Fréquent |
Peu fréquent |
|
Affections gastro-intestinales |
||
|
Diarrhée |
Très fréquent |
Peu fréquent |
|
Vomissements |
Très fréquent |
Peu fréquent |
|
Constipation |
Très fréquent |
Peu fréquent |
|
Nausées |
Très fréquent |
Peu fréquent |
|
Stomatite6) |
Fréquent |
Peu fréquent |
|
Affections hépatobiliaires |
||
|
Augmentation des ASAT |
Très fréquent |
Fréquent |
|
Augmentation des ALAT |
Très fréquent |
Fréquent |
|
Augmentation de la bilirubinémie7) |
Très fréquent |
Fréquent |
|
Augmentation de la phosphatase alcaline |
Très fréquent |
Peu fréquent |
|
Lésion hépatique d'origine médicamenteuse8) |
Peu fréquent |
Peu fréquent |
|
Affections de la peau et du tissu sous-cutané |
||
|
Eruption cutanée9) |
Très fréquent |
Fréquent |
|
Photosensibilité |
Fréquent |
Peu fréquent |
|
Affections musculo- squelettiques et systémiques |
||
|
Myalgie10) |
Très fréquent |
Peu fréquent |
|
Augmentation du taux sanguin de créatine phosphokinase |
Très fréquent |
Fréquent |
|
Affections du rein et des voies urinaires |
||
|
Lésion rénale aiguë |
Peu fréquent |
Peu fréquent** |
|
Augmentation de la créatininémie |
Fréquent |
Peu fréquent** |
|
Troubles généraux et anomalies au site d'administration |
||
|
Œdème11) |
Très fréquent |
Peu fréquent |
|
Investigations |
||
|
Augmentation du poids |
Très fréquent |
Peu fréquent |
|
Troubles du métabolisme et de la nutrition |
||
|
Hyperuricémie12) |
Fréquent |
-* |
*
Aucun effet indésirable de grade 3-4 n'a été observé.
** Comprend un événement de grade 5 (observé dans le CBNPC avancé).
1) comprend des cas d'anémie, de diminution de l'hémoglobine et
d'anémie normochrome normocytaire.
2) des cas rapportés dans l'étude BO40336 (N=128).
3) comprend des cas de dysgueusie, d'hypogueusie et de trouble du goût.
4) comprend des cas de vision trouble, d'atteinte visuelle, de corps
flottants vitréens, de diminution de l'acuité visuelle, d'asthénopie,
de diplopie, de photophobie et de photopsie.
5) comprend des cas de bradycardie et de bradycardie sinusale.
6) comprend des cas de stomatite et d'ulcération buccale.
7) comprend des cas d'augmentation du taux sanguin de bilirubine, d'hyperbilirubinémie, d'augmentation du taux sanguin de
bilirubine conjuguée et d'augmentation du taux sanguin de bilirubine non
conjuguée.
8) comprend deux patients avec une lésion hépatique d'origine
médicamenteuse selon le terme MedDRA ainsi qu'un
patient avec une augmentation des ALAT et ASAT de Grade 4 qui a une lésion
hépatique d'origine médicamenteuse documentée par une biopsie hépatique.
9) comprend des cas d'éruption cutanée, d'éruption cutanée maculopapuleuse, de dermatite acnéiforme, d'érythème,
d'éruption cutanée généralisée, d'éruption cutanée papuleuse, d'éruption
cutanée prurigineuse, d'éruption cutanée maculeuse, d'éruption cutanée
exfoliative et d'éruption érythémateuse.
10) comprend des cas de myalgie, de douleur musculo-squelettique et
d'arthralgie.
11) comprend des cas d'œdème périphérique, d'œdème, d'œdème
généralisé, d'œdème palpébral, d'œdème périorbital,
d'œdème facial, d'œdème localisé, de gonflement périphérique, de gonflement du
visage, de gonflement de la lèvre, de gonflement, de gonflement articulaire et
de gonflement de la paupière.
12) comprend des cas d'hyperuricémie et
d'augmentation de l'acide urique sanguin.
Description d'effets indésirables spécifiques
Pneumopathie interstitielle diffuse
Dans les essais cliniques, des cas de pneumopathie interstitielle diffuse sont survenus chez 1,3 % des patients traités par Alecensa, 0,4 % de ces cas étaient de grade 3 et des arrêts de traitement dûs à une pneumopathie interstielle diffuse sont survenus chez 0,9 % des patients. Dans l'essai clinique de phase III BO28984, aucun cas de pneumopathie interstitielle diffuse de Grade 3 ou 4 n'a été observé chez les patients traités par Alecensa versus 2,0 % des patients traités par le crizotinib. Il n'y a eu aucun cas de pneumopathie interstitielle diffuse d'issue fatale dans aucun des essais cliniques. Les patients doivent faire l'objet d'une surveillance des symptômes pulmonaires évocateurs d'une pneumopathie (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Hépatotoxicité
Dans les essais cliniques, trois patients ont présenté une lésion hépatique d'origine médicamenteuse documentée (dont deux patients pour lesquels l'effet indésirable rapporté était une lésion hépatique d'origine médicamenteuse et un patient présentant une augmentation des ASAT et ALAT de grade 4 dont l'analyse de la biopsie hépatique a conclu à une lésion hépatique d'origine médicamenteuse). Des augmentations des taux d'ASAT et d'ALAT ont été rapportées respectivement chez 22,7 % et 20,1 % des patients traités par Alecensa dans les essais cliniques. La majorité de ces événements était de Grade 1 et 2, et des événements de Grade ≥ 3 ont été rapportés chez 3,0 % des patients pour une augmentation des taux d'ASAT et 3,2 % des patients pour une augmentation des taux d'ALAT. Les événements sont généralement survenus au cours des trois premiers mois de traitement, ils étaient le plus souvent transitoires et réversibles à l'interruption temporaire du traitement par Alecensa (rapporté chez 2,3 % et 3,6 % des patients, respectivement) ou à une réduction de la posologie (1,7 % et 1,5 %, respectivement). Des augmentations des taux d'ASAT et d'ALAT, chez 1,1 % et 1,3 % des patients respectivement, ont conduit à l'arrêt du traitement par Alecensa. Des augmentations des taux d'ALAT ou d'ASAT de Grade 3 ou 4 ont chacune été observées chez 5 % des patients traités par Alecensa versus 16 % et 11 % des patients traités par crizotinib dans l'essai clinique de phase III BO28984.
Une augmentation du taux de bilirubine a été rapportée chez 25,1 % des patients traités par Alecensa dans les essais cliniques. La majorité des événements était d'intensité de Grade 1 et 2 ; des événements de Grade ≥ 3 ont été rapportés chez 3,4 % des patients. Ces événements sont généralement survenus au cours des trois premiers mois de traitement, ils étaient le plus souvent transitoires et la plupart étaient réversibles après modification de la posologie. Chez 7,7 % des patients, l'augmentation du taux de bilirubine a conduit à des modifications de posologie et chez 1,5 % des patients, l'augmentation du taux de bilirubine a conduit à l'arrêt du traitement par Alecensa. Dans l'essai clinique de phase III BO28984, une augmentation du taux de bilirubine de Grade 3 ou 4 est survenue chez 3,9 % des patients traités par Alecensa versus aucun patient traité par le crizotinib.
Une augmentation concomitante des ALAT ou ASAT supérieures ou égales à trois fois la LSN et de la bilirubine totale supérieure ou égale à deux fois la LSN, avec des phosphatases alcalines normales, a été rapportée chez un patient (0,2 %) traité dans les essais cliniques avec Alecensa.
Les patients doivent faire l'objet d'une surveillance de la fonction hépatique y compris les ALAT, ASAT et la bilirubine totale tel que décrit en rubrique Mises en garde spéciales et précautions d'emploi et doivent être pris en charge tel que recommandé en rubrique Posologie et mode d'administration.
Bradycardie
Des cas de bradycardie (11,1 %) de Grade 1 ou 2 ont été rapportés chez des patients traités par Alecensa au cours d'essais cliniques. Aucun patient n'a présenté d'événements de Grade ≥ 3. Cent- deux des 521 patients (19,6 %) traités par Alecensa, pour lesquels des ECG en série étaient disponibles, ont eu des valeurs de fréquence cardiaque post-dose inférieures à 50 battements par minute (bpm). Dans l'essai clinique de phase III BO28984, 15 % des patients traités par Alecensa ont eu des valeurs de fréquence cardiaque inférieures à 50 bpm versus 21 % des patients traités par le crizotinib. Les patients développant une bradycardie symptomatique doivent être pris en charge tel que recommandé en rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi. Aucun cas de bradycardie n'a entrainé l'arrêt du traitement par Alecensa.
Myalgie sévère et augmentation des CPK
Des cas de myalgie (34,9 %) comprenant des événements de myalgie (24,0 %), d'arthralgie (16,1 %) et des douleurs musculo-squelettiques (0,9 %) ont été rapportés chez des patients traités par Alecensa au cours des essais cliniques. La majorité des événements était de Grade 1 ou 2 et cinq patients (0,9 %) ont présenté un événement de Grade 3. Compte-tenu de ces effets indésirables, des modifications de la posologie du traitement par Alecensa ont été nécessaires pour neuf patients (1,7 %). Alecensa n'a pas été arrêté en raison de ces événements de myalgie. Une augmentation des CPK a été rapportée chez 55,6 % des 491 patients des essais cliniques pour lesquels des données biologiques sur les CPK étaient disponibles avec Alecensa. L'incidence d'une augmentation des CPK de Grade ≥ 3 était de 5,5 %. Le délai médian de survenue de l'augmentation des CPK de Grade ≥ 3 était de 15 jours dans les essais. Des modifications de dose suite à une augmentation des CPK ont été faites chez 5,3 % des patients ; aucun arrêt de traitement par Alecensa n'est survenu suite à des élévations des CPK. Dans l'essai clinique BO28984, une arthralgie sévère a été rapportée chez un patient (0,7 %) dans le bras alectinib et chez deux patients (1,3 %) dans le bras crizotinib. Une augmentation des CPK de Grade ≥ 3 a été rapportée chez 3,9 % des patients traités par Alecensa et 3,3 % des patients traités par crizotinib.
Anémie hémolytique
Une anémie hémolytique a été observée chez 3,1 % des patients traités par Alecensa au cours des essais cliniques. Ces cas étaient de grade 1 ou 2 (non graves) et n'ont pas conduit à l'arrêt du traitement (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Effets gastro-intestinaux
Des constipations (38,6 %), des nausées (17,4 %), des diarrhées (17,4 %) et des vomissements (12 %) étaient les effets gastro-intestinaux (GI) les plus fréquemment rapportés. La plupart de ces événements était de sévérité faible ou modérée ; des événements de Grade 3 ont été rapportés pour les diarrhées (0,9 %), les nausées (0,4 %), les vomissements (0,2 %) et les constipations (0,4 %). Ces événements n'ont pas conduit à l'arrêt du traitement par Alecensa. Le délai médian de survenue des événements de constipations, nausées, diarrhées et/ou vomissements dans les essais cliniques était de 21 jours. La fréquence de ces effets a diminué après le premier mois de traitement. Dans l'essai clinique de phase III BO28984, des événements de Grade 3 et 4 pour les nausées, diarrhées et constipations ont chacun été rapportés chez un patient (0,7 %) dans le bras alectinib et l'incidence des événements de Grade 3 et 4 pour les nausées, vomissements et diarrhées était respectivement de 3,3 %, 2,0 % et 3,3 % dans le bras crizotinib.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT L'INSTAURATION du traitement :
- Etablir le statut ALK-positif du CBNPC par une méthode d'analyse validée.
SURVEILLANCE du traitement :
- Symptôme pulmonaire évocateur d'une pneumopathie.
- Fonction hépatique : ALAT, ASAT, bilirubine totale à l'initiation du traitement puis toutes les 2 semaines pendant les 3 premiers mois de traitement; surveillance périodique par la suite.
- Taux de CPK toutes les 2 semaines pendant le premier mois de traitement puis lorsqu'il est cliniquement indiqué chez des patients déclarant des symptômes.
INFORMER les patients :
- Ils doivent déclarer toute douleur, sensibilité ou faiblesse musculaire inexpliquées.- Des signes et symptômes de perforations gastro-intestinales.
PREVENIR IMMEDIATEMENT LE MEDECIN en cas de :
- Douleurs sévères au niveau de l'estomac ou de l'abdomen, fièvre, frissons, nausées, vomissements ou rigidité abdominale ou ballonnements.
- Coloration jaune de la peau ou du blanc des yeux, douleur à droite de l'estomac, urines noires, démangeaisons de la peau, sensation de faim diminuée, nausées ou vomissements, sensation de fatigue, saignements ou apparition de bleus plus importante que normalement.
- Evanouissements, vertiges, pression sanguine basse, rythme cardiaque ralenti.
- Apparition de nouveaux symptômes pulmonaires ou aggravation des symptômes existants notamment difficulté à respirer, essoufflement, toux avec ou sans sécrétion, ou fièvre.
- Douleur inexpliquée des muscles ou douleurs musculaires qui ne disparaissent pas, sensibilités ou faiblesses musculaires.
- Sensation de fatigue, faiblesse ou essoufflement.
CONTRACEPTION DES FEMMES EN AGE DE PROCREER : Utiliser des méthodes contraceptives hautement efficaces pendant le traitement et pendant au moins 5 semaines suivant l'arrêt du traitement.
CONTRACEPTION CHEZ L'HOMME ayant des partenaires en âge de
procréer : Utiliser
des méthodes contraceptives hautement efficaces pendant le traitement et
pendant au moins 3 mois après l'arrêt du traitement. CONTACTER LE MEDECIN en cas de grossesse de la partenaire pendant cette période.
NE PAS S'EXPOSER au soleil pendant le traitement et jusqu'à au moins 7 jours après l'arrêt du traitement. Appliquer un écran solaire à large spectre anti-Ultraviolet A (UVA) / Ultraviolet B (UVB) et un baume pour les lèvres (Indice de protection solaire [SPF] ≥ 50).
PREVENIR LE MEDECIN en cas de prise de médicament à base de plantes contenant du millepertuis (Hypericum perforatum).
EVITER de consommer des oranges amères, des pamplemousses ou du jus de pamplemousse pendant le traitement.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (troubles de la vision, évanouissements, vertiges).
Femmes en âge de procréer
Les femmes en âge de procréer doivent être incitées à ne pas débuter une grossesse au cours de leur traitement par Alecensa (voir rubrique Mises en garde spéciales et précautions d'emploi).
Contraception chez les femmes traitées par Alecensa
Les patientes en âge de procréer recevant Alecensa doivent utiliser des méthodes contraceptives hautement efficaces durant le traitement et pendant au moins 5 semaines après l'arrêt du traitement par Alecensa (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Contraception chez les hommes traités par Alecensa
Les patients de sexe masculin ayant des partenaires en âge de procréer doivent utiliser des méthodes contraceptives hautement efficaces durant le traitement et pendant au moins 3 mois après l'arrêt du traitement par Alecensa (voir rubrique Mises en garde spéciales et précautions d'emploi).
Grossesse
Il n'y a pas ou peu de données issues de l'utilisation d'alectinib chez les femmes enceintes. D'après son mécanisme d'action, alectinib peut entraîner des malformations fœtales en cas d'administration chez une femme enceinte. Les études chez l'animal ont montré une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Les patientes, enceintes en cours de traitement par Alecensa ou au cours des 5 semaines suivant l'arrêt du traitement par Alecensa, doivent contacter leur médecin et être informées du risque potentiel pour le fœtus.
Les patients de sexe masculin dont les partenaires deviennent enceintes pendant la prise de traitement d'Alecensa, ou au cours des 3 mois qui suivent l'arrêt du traitement par Alecensa, doivent contacter leur médecin, et leur partenaire féminine doit consulter un médecin compte-tenu du risque potentiel pour le fœtus en raison de son potentiel aneugène (voir rubrique Données de sécurité préclinique).
Allaitement
On ne sait pas si l'alectinib et/ou ses métabolites sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut pas être exclu. Les mères doivent être averties qu'elles ne doivent pas allaiter pendant leur traitement par Alecensa.
Fertilité
Aucune étude chez l'animal n'a été réalisée pour évaluer l'effet d'alectinib sur la fertilité. Aucun effet indésirable sur les organes reproducteurs mâle ou femelle n'a été observé dans les études toxicologiques générales (voir rubrique Données de sécurité préclinique).
Effets des autres médicaments sur alectinib
D'après les données obtenues in vitro, le CYP3A4 est la
principale enzyme servant de médiateur sur le métabolisme à la fois
d'alectinib et de son principal métabolite actif (M4) et le CYP3A
contribue à 40- 50 % du métabolisme hépatique total. Le métabolite M4 a
montré une puissance et une activité similaire in vitro contre ALK.
Inducteurs du CYP3A
L'administration concomitante, une fois par jour, de doses orales multiples de 600 mg de rifampicine, un inducteur puissant du CYP3A, avec une dose orale unique de 600 mg d'alectinib a diminué la Cmax et l'ASCinf d'alectinib de respectivement 51 % et 73 % et a augmenté la Cmax et l'ASCinf du métabolite M4 de respectivement 2,20 et 1,79 fois. L'effet sur l'exposition associée à alectinib et au métabolite M4 était mineur, réduisant la Cmax et l'ASCinf de respectivement 4 % et 18 %. D'après les effets sur l'exposition
associée à alectinib et au métabolite M4, aucune adaptation posologique
n'est nécessaire en cas d'administration concomitante d'Alecensa et
d'inducteurs du CYP3A. Une surveillance appropriée est recommandée pour
les patients prenant des inducteurs puissants du CYP3A (incluant
notamment la carbamazépine, le phénobarbital, la phénytoïne, la
rifabutine, la rifampicine et le millepertuis).
Inhibiteurs du CYP3A
L'administration concomitante, deux
fois par jour, de doses orales multiples de 400 mg de posaconazole, un
inhibiteur puissant du CYP3A, avec une dose orale unique de 300 mg
d'alectinib a augmenté la Cmax et l'ASCinf d'alectinib de respectivement 1,18 et 1,75 fois, et a réduit la Cmax et l'ASC du métabolite M4 de respectivement 71 % et 25 %. L'effet sur l'exposition associée à alectinib et au métabolite M4 était mineur, réduisant la Cmax de 7 % et augmentant l'ASCinf de 1,36 fois. D'après
les effets sur l'exposition associée à alectinib et au métabolite M4,
aucune adaptation posologique n'est nécessaire en cas d'administration
concomitante d'Alecensa et d'inhibiteurs du CYP3A. Une surveillance
appropriée est recommandée pour les patients prenant des inhibiteurs
puissants du CYP3A (incluant notamment le ritonavir, le saquinavir, la
télithromycine, le kétoconazole, l'itraconazole, le voriconazole, le
posaconzaole, le néfazodone, le pamplemousse ou les oranges amères).
Médicaments augmentant le pH gastrique
Des doses multiples d'ésoméprazole, un inhibiteur de pompe à protons,
40 mg une fois par jour, n'a démontré aucun effet cliniquement
significatif sur l'exposition associée à alectinib et au métabolite M4.
Par conséquent, aucune adaptation posologique n'est nécessaire en cas
d'administration concomitante d'Alecensa et d'inhibiteurs de la pompe à
protons ou autres médicaments augmentant le pH gastrique (par ex : les
antagonistes du récepteur H2 ou antiacides).
Effet des transporteurs sur la disposition d'alectinib
Le métabolite M4 est un substrat de la glycoprotéine P (P-gp). Du fait
qu'alectinib inhibe la P-gp, il n'est pas attendu que l'administration
concomitante avec un inhibiteur de la P-pg ait un effet pertinent sur
l'exposition au métabolite M4.
Effets de l'alectinib sur d'autres médicaments
Les substrats du CYP
In vitro,
alectinib et son métabolite M4 montrent une faible inhibition
temps-dépendant du CYP3A4 et alectinib présente une faible induction
potentielle du CYP3A4 et du CYP2B6 à des concentrations cliniques.
Des
doses multiples de 600 mg d'alectinib n'avaient aucune influence sur
l'exposition du midazolam (2 mg), un substrat sensible du CYP3A. Par
conséquent, aucune adaptation posologique n'est nécessaire en cas de
co-administration avec des substrats du CYP3A.
Un risque d'induction du CYP2B6 et d'enzymes régulées par le pregnane X
receptor (PXR) excepté le CYP3A4 ne peut pas être complétement exclu.
L'efficacité d'une administration concomitante de contraceptifs oraux
peut être réduite.
Les substrats de la P-gp
In vitro,
alectinib et son principal métabolite actif M4 sont des inhibiteurs du
transporteur d'efflux P- gp. Par conséquent, alectinib et son
métabolite M4 peuvent avoir le potentiel d'augmenter les concentrations
plasmatiques des substrats de la P-gp co-administrés. Une surveillance
appropriée est recommandée en cas d'administration concomitante
d'alectinib avec des substrats de la P-gp (par ex : la digoxine, le
dabigatran etexilate, le topotécan, le sirolimus, l'évérolimus, le
nilotinib et le lapatinib).
Les substrats de la protéine de résistance du cancer du sein (BCRP)
In vitro,
alectinib et son métabolite M4 sont des inhibiteurs du transporteur
d'efflux BCRP. Par conséquent, alectinib et son métabolite M4 peuvent
avoir le potentiel d'augmenter les concentrations plasmatiques des
substrats de la BCRP co-administrés. Une surveillance appropriée est
recommandée en cas d'administration concomitante d'alectinib avec des
substrats de la BCRP (par ex : le méthotrexate, le mitoxantrone, le
topotécan et le lapatinib).
Le traitement par Alecensa doit être instauré et supervisé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Une méthode d'analyse d'ALK validée est nécessaire pour sélectionner les patients présentant un CBNPC avec réarrangement du gène ALK (ALK-positif). Le statut ALK-positif du CBNPC doit être établi avant l'instauration du traitement par Alecensa.
Posologie
La posologie recommandée d'Alecensa est de 600 mg (4 gélules de 150 mg) deux fois par jour au cours d'un repas (posologie quotidienne totale de 1200 mg).
Chez les patients atteints d'insuffisance hépatique sous-jacente sévère (Child-Pugh C), la posologie initiale recommandée est de 450 mg deux fois par jour au cours d'un repas (posologie quotidienne totale de 900 mg).
Durée de traitement
Traitement adjuvant du CBNPC réséqué
Le traitement par Alecensa doit être poursuivi jusqu'à récidive de la maladie, survenue d'une toxicité inacceptable ou pendant 2 ans.
Traitement du CBNPC avancé
Le traitement par Alecensa doit être poursuivi jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable.
Retard ou omission d'une dose
En cas d'omission d'une prise d'Alecensa, la dose omise doit être prise immédiatement sauf s'il reste moins de 6 heures avant la prochaine dose. Les patients ne doivent pas prendre deux doses en même temps pour compenser une dose omise. En cas de vomissement suite à une dose d'Alecensa, les patients doivent prendre la prochaine dose telle que planifiée.
Adaptations posologiques
La gestion des événements indésirables peut nécessiter une réduction de la posologie, une interruption temporaire ou un arrêt de traitement par Alecensa. La posologie d'Alecensa doit être réduite par palier de 150 mg deux fois par jour en fonction de la tolérance. Le traitement par Alecensa doit être définitivement arrêté en cas d'intolérance à la dose de 300 mg deux fois par jour.
Les recommandations d'adaptation de la posologie sont décrites dans les Tableaux 1 et 2 ci-dessous.
Tableau 1 Schéma de réduction de la posologie
|
Schéma de réduction de la posologie |
Palier de dose |
|
Posologie |
600 mg deux fois par jour |
|
Première réduction de la posologie |
450 mg deux fois par jour |
|
Deuxième réduction de la posologie |
300 mg deux fois par jour |
Tableau 2 Recommandations d'adaptation de la posologie en cas d'effets indésirables spécifiques (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables)
|
Grade CTCAE |
Traitement par Alecensa |
|
Pneumopathie interstitielle diffuse / pneumopathie de tout grade de sévérité |
Arrêter immédiatement et définitivement le traitement par Alecensa si aucune autre cause de maladie pulmonaire interstitielle / pneumopathie n'a été identifiée. |
|
Elévation du taux d'ALAT ou d'ASAT > 5 fois la LSN avec un taux de bilirubine totale ≤ 2 fois la LSN |
Interrompre temporairement le traitement jusqu'au retour à la valeur de référence ou ≤ 3 fois la LSN, puis reprendre le traitement au palier de dose inférieur (voir Tableau 1). |
|
Elévation du taux d'ALAT ou d'ASAT > 3 fois la LSN accompagnée d'une élévation du taux de bilirubine totale > 2 fois la LSN en l'absence de cholestase ou d'hémolyse |
Arrêter définitivement le traitement par Alecensa. |
|
Bradycardiea de Grade 2 ou de Grade 3 (symptomatique, potentiellement sévère et médicalement significative, nécessitant une intervention médicale) |
Interrompre
temporairement le traitement par Alecensa jusqu'au
retour à une bradycardie de Grade ≤ 1 (asymptomatique) ou à une
fréquence cardiaque ≥ 60 bpm. Evaluer les
médicaments concomitants connus pour entraîner une bradycardie, y compris les
médicaments antihypertenseurs. |
|
Bradycardiea de Grade 4 (conséquences menaçant le pronostic vital, nécessitant une intervention urgente) |
Arrêter
définitivement le traitement par Alecensa si aucun
médicament concomitant favorisant la bradycardie n'est identifié. |
|
Elévation des CPK > 5 fois la LSN |
Interrompre temporairement le traitement par Alecensa jusqu'au retour à la valeur de référence ou à un taux ≤ 2,5 fois la LSN, puis reprendre le traitement au même palier de dose. |
|
Elévation des CPK > 10 fois la LSN ou deuxième élévation des CPK > 5 fois la LSN |
Interrompre temporairement le traitement par Alecensa jusqu'au retour à la valeur de référence ou à un taux ≤ 2,5 fois la LSN, puis reprendre le traitement au palier de dose inférieur comme indiqué dans le Tableau 1. |
|
Anémie hémolytique avec un taux d'hémoglobine < 10 g/dL (Grade ≥ 2) |
Interrompre temporairement le traitement par Alecensa jusqu'à résolution, puis reprendre le traitement au palier de dose inférieur (voir Tableau 1). |
ALAT = alanine aminotransférase,
ASAT = aspartate aminotransférase, CPK = créatinine phosphokinase, CTCAE = National Cancer Institute (NCI) Common
Terminology Criteria for
Adverse Events, LSN = limite supérieure de la normale.
aFréquence cardiaque inférieure à 60
battements par minute (bpm).
Populations particulières
Insuffisance hépatique
Aucune adaptation de la posologie initiale n'est nécessaire chez les patients atteints d'insuffisance hépatique sous-jacente légère (Child-Pugh A) ou modérée (Child-Pugh B). La posologie initiale recommandée chez les patients atteints d'une insuffisance hépatique sous-jacente sévère (Child-Pugh C) est de 450 mg deux fois par jour (posologie quotidienne totale de 900 mg) (voir rubrique Propriétés pharmacocinétiques). Pour tous les patients atteints d'insuffisance hépatique, une surveillance appropriée (par exemple, marqueurs de la fonction hépatique) est recommandée (voir rubrique Mises en garde spéciales et précautions d'emploi).
Insuffisance rénale
Aucune adaptation posologique particulière n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. Alecensa n'a pas été étudié chez les patients atteints d'insuffisance rénale sévère. Cependant, l'élimination de l'alectinib par voie rénale étant négligeable, aucune adaptation posologique n'est nécessaire chez les patients atteints d'insuffisance rénale sévère (voir rubrique Propriétés pharmacocinétiques).
Patients âgés (≥ 65 ans)
Les données limitées sur la tolérance et l'efficacité d'Alecensa chez les patients âgés de 65 ans et plus ne suggèrent pas qu'une adaptation posologique soit nécessaire pour les patients âgés (voir rubrique Propriétés pharmacocinétiques). Aucune donnée n'est disponible pour les patients âgés de plus de 80 ans.
Population pédiatrique
La sécurité et l'efficacité d'Alecensa chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Patients de très haut poids corporel (> 130 kg)
Bien que les simulations de pharmacocinétique (PK) sur Alecensa n'indiquent pas une faible exposition chez les patients de très haut poids corporel (> 130 kg), alectinib est largement distribué et les études cliniques sur alectinib ont inclus des patients de poids corporel entre 36,9 et 123 kg. Aucune donnée n'est disponible pour les patients de poids corporel supérieur à 130 kg.
Mode d'administration
Alecensa est destiné à une administration orale. Les gélules doivent être avalées entières et ne doivent pas être ouvertes ou dissoutes. Elles doivent être prises au cours d'un repas (voir rubrique Propriétés pharmacocinétiques).
Durée de conservation :
5 ans.
Précautions particulières de conservation :
Plaquettes thermoformées
A conserver dans l'emballage d'origine à l'abri de l'humidité.
Sans objet.
En cas de surdosage, les patients doivent être étroitement surveillés et des soins symptomatiques généraux instaurés. Il n'existe aucun antidote spécifique en cas de surdosage à Alecensa.
Classe pharmacothérapeutique: agent antinéoplasique, inhibiteur de protéine kinase, Code ATC: L01ED03.
Mécanisme d'action
Alectinib est un inhibiteur de tyrosine kinase ALK et RET (REarranged during Transfection) hautement sélectif et puissant. Dans les études précliniques, l'inhibition de l'activité tyrosine kinase de l'ALK a conduit au blocage en aval des voies de signalisation incluant le transducteur du signal et l'activateur de transcription 3 (STAT3) et la phosphoinositide 3-kinase (PI3K)/protéine kinase B (AKT) et à l'induction de la mort cellulaire tumorale (apoptose).
Alectinib a démontré une activité in vitro et in vivo contre les formes mutantes de l'enzyme ALK, dont les mutations responsables de la résistance au crizotinib. Le principal métabolite de l'alectinib (M4) a montré une activité et une puissance in vitro similaires.
D'après les données précliniques, l'alectinib n'est pas un substrat de la P-gp ou de la BCRP, chacun étant des transporteurs d'efflux au niveau de la barrière hémato-encéphalique, et est par conséquent en mesure de distribuer et d'être retenu au sein du système nerveux central.
Efficacité et sécurité clinique
Traitement adjuvant du CBNPC réséqué ALK-positif
L'efficacité d'Alecensa dans le traitement adjuvant des patients atteints d'un CBNPC ALK-positif après résection complète de la tumeur a été établie dans un essai clinique international randomisé de phase III, en ouvert (BO40336, ALINA). Les patients éligibles devaient avoir un CBNPC de stade IB (tumeurs ≥ 4 cm) - Stade IIIA selon le système de stadification de l'Union Internationale Contre le Cancer / American Joint Committee on Cancer (UICC / AJCC), 7ème édition, avec une maladie ALK-positive identifiée par un test ALK local marqué CE, ou un test central réalisé avec le kit d'immunohistochimie (IHC) Ventana anti-ALK (D5F3).
Les critères de sélection suivants définissent les patients avec un risque élevé de récidive qui sont inclus dans l'indication thérapeutique et reflètent la population de patients atteints d'un CBNPC de stade IB (tumeurs ≥ 4 cm) - IIIA selon la 7ième édition du système de stadification de l'UICC / AJCC :
Taille de la tumeur ≥ 4 cm ; ou des tumeurs de toute taille associées à un statut N1 ou N2 ; ou des tumeurs invasives des structures thoraciques (envahissant directement la plèvre pariétale, la paroi thoracique, le diaphragme, le nerf phrénique, la plèvre médiastinale, le péricarde pariétal, le médiastin, le cœur, les grands vaisseaux, la trachée, le nerf laryngé récurrent, l'œsophage, le corps vertébral, la carène) ; ou des tumeurs atteignant la bronche principale distantes de < 2 cm de la carène mais sans atteinte de celle-ci ; ou des tumeurs associées à une atélectasie ou à une pneumopathie obstructive du poumon entier ; ou des tumeurs avec un(des) nodule(s) séparé(s) dans le même lobe ou dans un lobe ipsilatéral différent de la tumeur primitive.
L'essai
n'a pas inclus de patients présentant un statut N2 avec des tumeurs envahissant
le médiastin, le cœur, les grands vaisseaux, la trachée, le nerf laryngé
récurrent, l'œsophage, le corps vertébral, la carène, ou avec un(des) nodule(s)
tumoral(aux) séparé(s) dans un lobe ipsilatéral
différent.
Après résection de la tumeur, les patients ont été randomisés selon un rapport
1:1 pour recevoir Alecensa ou une chimiothérapie à
base de platine. La randomisation était stratifiée selon l'origine ethnique
(asiatique et non asiatique) et le stade de la maladie (IB, II et IIIA). Alecensa était administré par voie orale, à la posologie
recommandée de 600 mg deux fois par jour pendant une durée totale de 2 ans, ou
jusqu'à la récidive de la maladie ou d'une toxicité inacceptable. La
chimiothérapie à base de platine était administrée par voie intraveineuse
pendant 4 cycles, chaque cycle durant 21 jours, selon un des schémas
thérapeutiques suivants :
Cisplatine 75 mg/m2 le
Jour 1 plus vinorelbine 25 mg/m2 les jours
1 et 8 ;
Cisplatine 75 mg/m2 le Jour 1 plus gemcitabine 1 250 mg/m2 les jours 1 et 8 ;
Cisplatine 75 mg/m2 le Jour 1 plus pemetrexed 500 mg/m2 le jour 1.
En cas d'intolérance à un des protocoles à base de cisplatine, le carboplatine était administré à la place du cisplatine dans les associations ci-dessus à une dose d'aire sous la courbe (ASC) du carboplatine libre de 5 mg/mL/min ou 6 mg/mL/min.
Le critère principal d'efficacité était la survie sans maladie (DFS) évaluée par l'investigateur. La DFS était définie comme le délai entre la date de randomisation et la date d'apparition de l'un des évènements suivants : première récidive documentée de la maladie ; nouveau CBNPC primaire ; ou décès, toutes causes confondues, selon l'évènement survenant en premier. Les critères secondaires et exploratoires d'efficacité étaient la survie globale (OS) et le délai de récidive au niveau du système nerveux central (CNS) (DFS-SNC) ou décès.
Un total de 257 patients a été étudié : 130 patients ont été randomisés dans le bras Alecensa, et 127 patients ont été randomisés dans le bras chimiothérapie. Globalement, l'âge médian était de 56 ans (intervalle : 26 à 87), et 24 % étaient âgés de ≥ 65 ans, 52 % étaient des femmes, 56 % étaient asiatiques, 60 % n'avaient jamais fumé, 53 % avaient un indice de performance ECOG de 0, 10 % avaient un stade IB, 36 % avaient un stade II et 54 % avaient un stade IIIA.
ALINA a démontré une amélioration statistiquement significative de la DFS chez les patients traités par Alecensa par rapport aux patients traités par chimiothérapie dans les populations de patients de stade II - IIIA et de stade IB (≥ 4 cm) - IIIA (ITT). Les données d'OS n'étaient pas matures au moment de l'analyse de la DFS avec 2,3 % de décès rapportés dans l'ensemble. La durée médiane du suivi de survie était de 27,8 mois dans le bras Alecensa et de 28,4 mois dans le bras chimiothérapie.
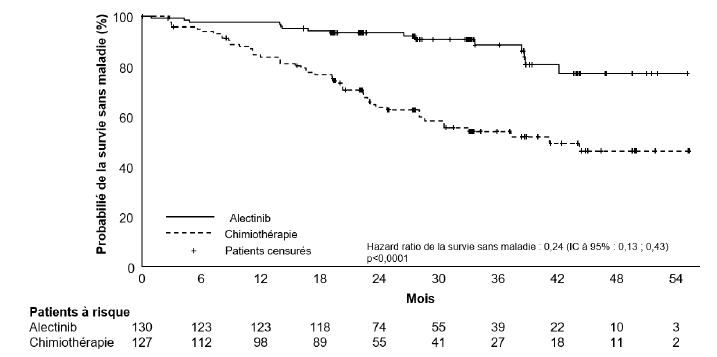
Les résultats d'efficacité de la DFS sont résumés dans le tableau 4 et la figure 1.
Tableau 4 Résultats de la DFS évalués par l'investigateur dans ALINA
| Critères d'efficacité | Stade II - IIIA | Population ITT | ||
|
Alecensa
N = 116 |
Chimiothérapie N = 115 |
Alecensa
N = 130 |
Chimiothérapie N = 127 |
|
|
Nombre d'événements de la DFS (%) |
14 (12,1) | 45 (39,1) | 15 (11,5) | 50 (39,4) |
|
Médiane
de la DFS (IC à 95 %) |
NE (NE ; NE) |
44,4 (27,8 ; NE) |
NE (NE ; NE) |
41,3 (28,5 ; NE) |
|
HR
stratifié (IC à 95 %)* |
0,24 (0,13 ; 0,45) |
0,24 (0,13 ; 0,43) |
||
|
Valeur
de p (log- rank)* |
< 0,0001 | < 0,0001 | ||
DFS =
survie sans maladie ; ITT = intention de traiter ; IC = intervalle de confiance
; NE = non évaluable ; HR = hazard ratio
* Stratifié selon l'origine ethnique dans le stade II-IIIA, stratifié selon l'origine ethnique et le stade IB-IIIA.
* Stratifié selon l'origine ethnique dans le stade II-IIIA, stratifié selon l'origine ethnique et le stade IB-IIIA.
Figure 1 : Courbe de Kaplan-Meier de la DFS évaluée par l'investigateur dans la population ITT
Traitement du CBNPC avancé ALK-positif
Patients naïfs de traitement
La sécurité et l'efficacité d'Alecensa ont été étudiées dans un essai clinique international randomisé de phase III, en ouvert (BO28984, ALEX) chez des patients naïfs de traitement atteints d'un CBNPC ALK-positif. Des tests centralisés évaluant la positivité de l'expression de la protéine ALK dans des échantillons de tissus de tous les patients par immunohistochimie avec le kit Ventana anti-ALK (D5F3), étaient requis avant la randomisation dans l'essai.
Un total de 303 patients a été inclus dans l'essai de phase III, 151 patients randomisés dans le bras crizotinib et 152 patients randomisés dans le bras Alecensa, recevant Alecensa par voie orale, à la posologie recommandée de 600 mg deux fois par jour.
L'indice de performance ECOG ((Eastern Cooperative Oncology Group) (0/1 vs. 2)), l'origine ethnique (asiatique vs. non asiatique) et les métastases cérébrales (SNC) à l'inclusion (oui vs. non) étaient des critères de stratification de la randomisation. Le critère principal d'évaluation de l'essai était de montrer la supériorité d'Alecensa versus crizotinib, basée sur la survie sans progression (SSP) selon l'évaluation de l'investigateur utilisant RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1. Les caractéristiques démographiques et pathologiques à l'inclusion dans le bras Alecensa étaient un âge médian de 58 ans (54 ans dans le bras crizotinib), 55 % de femmes (58 % dans le bras crizotinib), 55 % de non asiatiques (54 % dans le bras crizotinib), 61 % sans antécédent tabagique (65 % dans le bras crizotinib), 93 % ayant un indice de performance ECOG de 0 ou 1 (93 % dans le bras crizotinib), 97 % à un stade IV de la maladie (96 % dans le bras crizotinib), 90 % ayant une histologie d'adénocarcinome (94 % dans le bras crizotinib), 40 % ayant des métastases cérébrales à l'inclusion (38 % dans le bras crizotinib) et 17 % ayant précédemment reçu une radiothérapie au niveau cérébral (14 % dans le bras crizotinib).
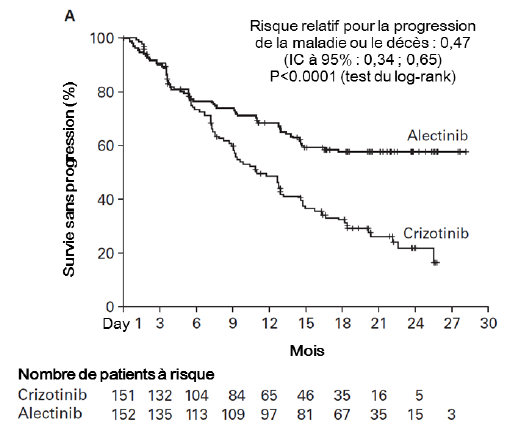
L'essai a atteint son critère principal lors de l'analyse primaire, montrant une amélioration statistiquement significative de la survie sans progression par l'investigateur. Les données d'efficacité sont résumées dans le Tableau 5 et la courbe de Kaplan-Meier de la survie sans progression évaluée par l'investigateur est représentée en Figure 2.
Tableau 5 Résumé des résultats d'efficacité de l'étude BO28984 (ALEX)
|
Crizotinib
N = 151 |
Alectinib
N = 152 |
|
| Durée médiane de suivi (mois) |
17,6 (limite 0,3 - 27,0) |
18,6 (limite 0,5 - 29,0) |
|
Critères
primaires d'efficacité
Survie Sans Progression (INV) Nombre
de patients ayant présenté un
événement n (%) Médiane (mois) [IC à 95 %] Risque Relatif (HR) [IC à 95 %] log rank p-value stratifié |
102 (68 %) 11,1 [9,1 ; 13,1] |
62 (41 %) NE [17,7 ; NE] |
|
0,47 [0,34 ; 0,65] p < 0,0001 |
||
|
Critères
secondaires d'efficacité
Survie Sans Progression (CRI) *Nombre
de patients ayant présenté un
événement n (%) Médiane (mois) [IC à 95 %] Risque Relatif (HR) [IC à 95 %] log rank p-value stratifié |
92 (61 %) 10,4 [7,7 ; 14,6] |
63 (41 %) 25,7 [19,9 ; NE] |
|
0,50 [0,36 ; 0,70] p < 0,0001 |
||
|
Temps
de progression cérébrale (CRI)*, **
Nombre
de patients ayant présenté un
événement n (%) Rapport de risque instantané cause-spécifique [IC à 95 %] log rank p-value stratifié |
68 (45 %) |
18 (12 %) |
|
0,16 [0,10 ; 0,28] |
||
|
Incidence cumulée à 12 mois de la progression cérébrale (CRI) en % [IC à 95 %] |
p < 0,0001 | |
|
41,4 % [33,2 ; 49,4] |
9,4 % [5,4 ; 14,7] |
|
|
Taux
de réponse objectif (INV)*, ***
Répondeurs
n (%)
[IC à 95 %] |
114 (75,5 %) [67,8 ; 82,1] |
126 (82,9 %) [76,0 ; 88,5] |
|
Survie
globale*
Nombre
de patients ayant présenté un
événement N (%) Médiane (mois) [IC à 95 %] Risque Relatif (HR) [IC à 95 %] |
40 (27 %) NE [NE ; NE] |
35 (23 %) NE [NE ; NE] |
|
0,76 [0,48 ; 1,20] |
||
|
Durée
de Réponse (INV)
Médiane
(mois)
[IC à 95 %] |
N = 114 11,1 [7,9 ; 13,0] |
N = 126 NE [NE ; NE] |
|
Taux
de réponse objectif cérébral chez des patients avec métastases cérébrales mesurables à l'inclusion Répondeurs
cérébraux n (%)
[IC à 95 %] Nombre de réponses cérébrales complètes (%) Durée de réponse cérébrale, médiane (mois) [IC à 95 %] |
N = 22 11 (50,0 %) [28,2 ; 71,8] 1 (5 %) 5,5 [2,1 ; 17,3] |
N = 21 17 (81,0 %) [58,1 ; 94,6] 8 (38 %) 17,3 [14,8 ; NE] |
|
Taux
de réponse objectif cérébral chez des patients avec métastases cérébrales mesurables et non mesurables à l'inclusion (CRI) Répondeurs
cérébraux n (%)
[IC à 95 %] Nombre de réponses cérébrales complètes (%) Durée de réponse cérébrale, médiane (mois) [IC à 95 %] |
N = 58 15 (25,9 %) [15,3 ; 39,0] 5 (9 %) 3,7 [3,2 ; 6,8] |
N = 64 38 (59,4 %) [46,4 ; 71,5] 29 (45 %) NE [17,3 ; NE] |
*
Principaux critères secondaires d'évaluation hiérarchisés
** Analyse de risque compétitive de la progression cérébrale, de la progression systémique et du décès en tant qu'événements compétitifs
*** 2 patients dans le bras crizotinib et 6 patients dans le bras alectinib ont eu une réponse complète
IC = intervalle de confiance ; CRI = comité de revue indépendant ; INV = investigateur ; NE = non estimable
** Analyse de risque compétitive de la progression cérébrale, de la progression systémique et du décès en tant qu'événements compétitifs
*** 2 patients dans le bras crizotinib et 6 patients dans le bras alectinib ont eu une réponse complète
IC = intervalle de confiance ; CRI = comité de revue indépendant ; INV = investigateur ; NE = non estimable
Le bénéfice de la survie sans progression était cohérent pour les patients avec des métastases cérébrales à l'inclusion (hazard ratio (HR) = 0,40, intervalle de confiance (IC) à 95 % [0,25 ; 0,64], survie sans progression médiane pour Alecensa = non estimable (NE), IC à 95 % [9,2 ; NE], survie sans progression médiane pour crizotinib = 7,4 mois, IC à 95 % [6,6 ; 6,9]) et sans métastase cérébrale à l'inclusion (HR = 0,51, IC à 95 % [0,33 ; 0,80], survie sans progression médiane pour Alecensa = NE, IC à 95 % [NE ; NE], survie sans progression médiane pour crizotinib = 14,8 mois, IC à95 % [10,8 ; 20,3]), indiquant un bénéfice d'Alecensa sur le crizotinib dans les deux sous-groupes.
Figure 2 : Courbe de Kaplan Meier de la survie sans progression évaluée par l'investigateur dans l'étude BO28984 (ALEX)
Patients pré-traités par crizotinib
La sécurité et l'efficacité d'Alecensa ont été étudiées chez des patients atteints de CBNPC ALK-positif, pré-traités par crizotinib, dans le cadre de deux essais cliniques de phase I/II (NP28673 et NP28761).
NP28673
L'étude NP28673 était une étude de phase I/II mono-bras, multicentrique,
conduite chez des patients atteints d'un CBNPC ALK-positif avancé et ayant
préalablement progressé après un traitement par crizotinib.
Outre le crizotinib, les patients pouvaient avoir
reçu un traitement préalable par chimiothérapie. Un total de 138 patients a été
inclus dans la partie phase II de l'étude et a reçu Alecensa
par voie orale, à la posologie recommandée de 600 mg deux fois par jour.
Le critère principal était d'évaluer l'efficacité d'Alecensa par le taux de réponse objective à partir de l'analyse centralisée du Comité de Revue Indépendant (CRI) utilisant les critères RECIST version 1.1 dans la population générale (avec ou sans exposition préalable à des traitements de chimiothérapie cytotoxique). Le co-critère primaire était d'évaluer le taux de réponse objective à partir de l'analyse centralisée du Comité de Revue Indépendante (CRI) utilisant les critères RECIST version 1.1 chez les patients ayant été préalablement exposés à des traitements de chimiothérapie cytotoxique. Si la borne inférieure de l'intervalle de confiance du taux de réponse objective estimée était au-dessus du seuil pré-spécifié de 35 % alors le résultat était considéré comme statistiquement significatif.
La démographie des patients était cohérente avec celle décrite dans la population des CBNPC ALK-positif. Les caractéristiques démographiques de la population globale de l'étude étaient constituées de 67 % de caucasiens, 26 % d'asiatiques, 56 % de femmes et l'âge médian était de 52 ans. La majorité des patients n'avait pas d'antécédent tabagique (70 %). L'indice de performance ECOG à l'initiation était de 0 ou 1 chez 90,6 % des patients et 2 chez 9,4 % des patients. Lors de l'inclusion dans l'étude, 99 % des patients présentaient une maladie de stade IV, 61 % présentaient des métastases cérébrales et 96 % des tumeurs étaient considérées comme des adénocarcinomes. Parmi les patients inclus dans l'étude, 20 % des patients avaient progressé après un traitement préalable par crizotinib et 80 % avaient progressé après un traitement par crizotinib et après au moins un traitement de chimiothérapie.
Etude
NP28761
L'étude NP28761 était une étude de phase I/II mono-bras, multicentrique,
conduite chez des patients atteints d'un CBNPC ALK-positif avancé et ayant
préalablement progressé après un traitement par crizotinib.
Outre le crizotinib, les patients pouvaient avoir
reçu un traitement préalable de chimiothérapie. Un total de 87 patients était
inclus dans la partie phase II de l'étude et a reçu Alecensa
par voie orale, à la posologie recommandée de 600 mg deux fois par jour.
Le critère principal était d'évaluer l'efficacité d'Alecensa par le taux de réponse objectif à partir de l'analyse centralisée du Comité de Revue Indépendant (CRI) utilisant les critères RECIST version 1.1. Si la borne inférieure de l'intervalle de confiance du taux de réponse objective estimée était au-dessus du seuil pré-spécifié de 35 % alors le résultat était considéré comme statistiquement significatif.
La démographie des patients était cohérente avec celle décrite dans la population des CBNPC ALK-positif. Les caractéristiques démographiques de la population globale de l'étude étaient constituées de 84 % de caucasiens, 8 % d'asiatiques, 55 % de femmes. L'âge médian était de 54 ans. La majorité des patients n'avait pas d'antécédent tabagique (62 %). L'indice de performance ECOG à l'initiation était de 0 ou 1 pour 89,7 % des patients et de 2 pour 10,3 % des patients. Lors de l'inclusion dans l'étude, 99 % des patients présentaient une maladie de stade IV, 60 % présentaient des métastases cérébrales et 94 % des tumeurs des patients étaient considérées comme des adénocarcinomes. Parmi les patients inclus dans l'étude, 26 % des patients avaient progressé après un traitement préalable par crizotinib et 74 % avaient progressé après un traitement par crizotinib et après au moins un traitement de chimiothérapie.
Les résultats principaux des études NP28673 et NP28761 sont résumés dans le Tableau 6. Un résumé de l'analyse poolée des critères SNC est présenté dans le Tableau 7.
Tableau 6 : Résultats d'efficacité des études NP28673 et NP28761
|
NP28673 Alecensa 600 mg Deux fois par jour |
NP28761 Alecensa 600 mg Deux fois par jour |
|
| Durée moyenne de suivi (mois) |
21 (limite 1 - 30) |
17 (limite 1 - 29) |
|
Critères
primaires d'efficacité
Taux de réponse objective (CRI) dans la population pour laquelle une réponse était évaluable Répondeurs
N (%)
[IC 95 %] Taux de réponse objective (CRI) chez les patients pré-traités par chimiothérapie Répondeurs
N (%)
[IC 95 %] |
N = 122 a 62 (50,8 %) [41,6 % ; 60,0 %] N = 96 43 (44,8 %) [34,6 %; 55,3 %] |
N = 67 b 35 (52,2 %) [39,7 %; 64,6 %] |
|
Critères
secondaires d'efficacité Durée de la réponse (CRI)
Nombre
de patients ayant
présenté un événement N (%) Médiane (mois) [IC
95 %]
Survie Sans Progression (CRI) Nombre
de patients ayant
présenté un événement N (%) Durée de la médiane (mois) [IC 95 %] |
N = 62 36 (58,1 %) 15,2 [11,2 ; 24,9] N = 138 98 (71,0 %) 8,9 [5,6 ; 12,8] |
N = 35 20 (57,1 %) 14,9 [6,9 ; NE] N = 87 58 (66,7) 8,2 [6,3; 12,6] |
IC
= intervalle de confiance ; CRI = comité de revue indépendant ; NE = non
estimable
a Selon le comité de revue indépendant, 16 patients ne présentaient pas de maladie mesurable à l'initiation et n'ont pas été inclus dans la population pour laquelle une réponse était évaluable.
b Selon le comité de revue indépendant, 20 patients ne présentaient pas de maladie mesurable à l'initiation et n'ont pas été inclus dans la population pour laquelle une réponse était évaluable.
a Selon le comité de revue indépendant, 16 patients ne présentaient pas de maladie mesurable à l'initiation et n'ont pas été inclus dans la population pour laquelle une réponse était évaluable.
b Selon le comité de revue indépendant, 20 patients ne présentaient pas de maladie mesurable à l'initiation et n'ont pas été inclus dans la population pour laquelle une réponse était évaluable.
Les résultats du taux de réponse objective des études NP28673 et NP28761 étaient cohérents dans tous les sous-groupes de patients caractérisés à l'initiation de l'étude, tels que l'âge, le sexe, l'ethnie, l'indice de performance ECOG, la présence de métastases cérébrales et le traitement préalable par chimiothérapie, notamment si l'on considère le faible nombre de patients dans quelques sous-groupes.
Tableau 7 Résumé de l'analyse poolée des critères SNC à partir des études NP28673 et NP28761
| Paramètres cérébraux (NP28673 et NP28761) | Alecensa 600 mg deux fois par jour |
|
Patients
porteurs de métastases cérébrales mesurables à l'inclusion Taux de réponse objective cérébrale (CRI) Répondeurs
(%)
[IC 95 %] Réponse Complète Réponse Partielle Durée de réponse objective cérébrale (CRI) Nombre
de patients ayant présenté un événement N (%)
Médiane (mois) [IC 95 %] |
N = 50 32 (64,0 %) [49,2 % ; 77,1 %] 11 (22,0 %) 21 (42,0 %) N = 32 18 (56,3 %) 11,1 [7,6 ; NE] |
IC =
intervalle de confiance ; CRI = comité de revue indépendante ; NE = non
estimable
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Alecensa dans tous les sous-groupes de la population pédiatrique dans le carcinome du poumon (carcinome à petites cellules et non à petites cellules) (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Les paramètres pharmacocinétiques pour alectinib et son principal métabolite actif (M4) ont été caractérisés chez les patients CBNPC ALK-positif et les sujets sains. Selon les analyses pharmacocinétiques de population, la moyenne géométrique (coefficient de variation %) de l'état d'équilibre de la Cmax, Cmin et ASC0-12hr pour alectinib était environ de 665 ng/ml (44,3 %), 572 ng/mL (47,8 %) et 7 430 ng*h/mL (45,7 %), respectivement. La moyenne géométrique de l'état d'équilibre de la Cmax, Cmin et ASC0-12hr pour le métabolite M4 était environ de 246 ng/mL (45,4 %), 222 ng/mL (46,6 %) et 2 810 ng*h/mL (45,9 %), respectivement.
Absorption
Après l'administration orale d'une dose de 600 mg deux fois par jour avec un
repas, chez des patients CBNPC ALK-positif, alectinib
était absorbé à un Tmax après 4 à 6 heures
environ.
L'état d'équilibre d'alectinib est atteint dans les 7 jours avec une administration continue de 600 mg deux fois par jour. Le rapport d'accumulation pour le régime posologique de 600 mg deux fois par jour était d'environ 6 fois. L'analyse pharmacocinétique de population soutient la proportionnalité de doses pour alectinib sur l'intervalle de doses allant de 300 à 900 mg à jeun.
La biodisponibilité absolue des gélules alectinib était de 36,9% (IC à 90 % : 33,9 % ; 40,3 %) avec un repas chez des sujets sains.
Après l'administration d'une dose orale unique de 600 mg avec un repas riche en graisses et en calories, l'exposition d'alectinib et du métabolite M4 a triplé par rapport à celle dans des conditions de jeûne (voir rubrique Posologie et mode d'administration).
Distribution
Alectinib et son principal métabolite M4 sont
fortement liés aux protéines plasmatiques humaines (> 99 %) indépendamment
de la concentration de la substance active. A des concentrations cliniquement
significatives, les rapports de concentration moyens sang/plasma in vitro d'alectinib et du métabolite M4 sont de 2,64 et 2,50
respectivement.
Le volume de distribution moyen géométrique à l'état d'équilibre (Vss) d'alectinib après administration IV était de 475 L, ce qui indique une large distribution tissulaire.
Selon les données in vitro, alectinib n'est pas un substrat de la P-gp. Alectinib et le métabolite M4 ne sont pas des substrats de la BCRP ou du polypeptide transporteur d'anions organiques (OATP) 1BI/B3.
Biotransformation
Des
études de métabolisme in vitro ont montré que le CYP3A4 était le
principal isoenzyme impliqué dans le métabolisme d'alectinib et de son principal métabolite M4. Sa
contribution est estimée à 40- 50% du métabolisme d'alectinib.
Les résultats de l'étude du bilan de masse humaine a
démontré qu'alectinib et le métabolite M4 étaient les
principaux agents circulant dans le plasma, avec 76 % de la radioactivité totale
dans le plasma. Le rapport de la moyenne géométrique métabolite/molécule mère à
l'état d'équilibre est de 0,399.
Le
métabolite M1b a été détecté en tant que métabolite mineur in vitro et
dans le plasma chez des sujets sains. La formation du métabolite M1b et de son
isomère mineur M1a serait probablement catalysée par une association d'isoenzymes du CYP (comprenant des isoenzymes
autres que le CYP3A) et des enzymes aldéhyde déshydrogénases.
Des études in vitro indiquent que ni alectinib ni son métabolite actif majeur (M4) n'inhibent les CYP1A2, CYP2B6, CYP2C9, CYP2C19 ou CYP2D6 à des concentrations cliniquement pertinentes. Alectinib n'inhibe pas les transporteurs OATP1B1/OATP1B3, OAT1, OAT3 ou OCT2 à des concentrations cliniquement pertinentes in vitro.
Élimination
Après
administration d'une dose unique d'alectinib marqué
au 14C administré par voie orale à des sujets sains, la majorité de
la radioactivité a été excrétée dans les fèces (Récupération moyenne : 97,8 %)
avec une excrétion minimale dans les urines (Récupération moyenne : 0,46 %).
Dans les fèces, 84 % et 5,8 % de la dose a été excrétée sous forme inchangée d'alectinib ou du métabolite M4, respectivement.
D'après
une analyse pharmacocinétique de population, la clairance apparente (CL/F) d'alectinib était de 81,9 L/heure. La moyenne géométrique des
estimations de demies-vie d'élimination individuelle pour l'alectinib
était de 32,5 heures. Les valeurs correspondantes pour le métabolite M4 étaient
217 L/heure et 30,7 heures, respectivement.
Pharmacocinétique dans des populations particulières
Insuffisance rénale
Des
quantités négligeables d'alectinib et du métabolite
actif M4 sont excrétés sous forme inchangée dans les urines (< 0,2 % de la
dose). D'après une analyse pharmacocinétique de population, les expositions à alectinib et au métabolite M4 étaient similaires chez les
patients présentant une insuffisance rénale légère à modérée et une fonction
rénale normale. La pharmacocinétique d'alectinib n'a
pas été étudiée chez les patients présentant une insuffisance rénale sévère.
Insuffisance hépatique
L'élimination
d'alectinib étant principalement métabolisé par le
foie, une insuffisance hépatique peut augmenter la concentration plasmatique d'alectinib et/ou son métabolite principal M4. D'après une
analyse pharmacocinétique de population, les expositions à alectinib
et au métabolite M4 étaient similaires chez les patients présentant une
insuffisance hépatique légère et une fonction hépatique normale.
Après administration d'une dose orale unique de 300 mg d'alectinib chez des patients atteints d'insuffisance hépatique sévère (Child-Pugh C), la Cmax d'alectinib était identique et l'ASCinf d'alectinib était 2,2 fois plus élevée par rapport aux mêmes paramètres chez des sujets sains appariés. La Cmax et l'ASCinf de son métabolite M4 ont été respectivement plus basses de 39 % et 34 %, conduisant à une exposition combinée à alectinib et à M4 (ASCinf) 1,8 fois plus élevée chez des patients atteints d'insuffisance hépatique sévère par rapport aux sujets sains appariés.
L'étude de l'insuffisance hépatique a également inclus un groupe de patients atteints d'insuffisance hépatique modérée (Child-Pugh B) et une exposition à alectinib modérement plus élevée a été observée dans ce groupe par rapport aux sujets sains appariés. Cependant, les patients du groupe Child-Pugh B n'ont généralement pas présenté d'anomalies de la bilirubine, de l'albumine ou du taux de prothrombine, suggérant qu'ils ne sont pas complétement représentatifs des patients atteints d'insuffisance hépatique modérée avec une capacité métabolique diminuée.
Effets de l'âge, du poids corporel, de l'origine ethnique
et du sexe
L'âge,
le poids corporel, l'origine ethnique et du sexe n'ont pas eu d'effet clinique
significatif sur l'exposition systémique à alectinib
et au métabolite M4. Les patients inclus dans les essais cliniques avaient un
poids corporel entre 36,9 et 123 kg. Aucune donnée n'est disponible pour les
patients de très haut poids corporel (> 130 kg) (voir rubrique Posologie
et mode d'administration).
Alecensa a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Une attention particulière doit être portée pendant la conduite de véhicules ou l'utilisation de machines, les patients pouvant présenter une bradycardie symptomatique (par ex : syncope, vertige, hypotension) ou des troubles visuels durant le traitement par Alecensa (voir rubrique Effets indésirables).
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée pour établir le potentiel carcinogène d'alectinib.
Mutagénicité
Alectinib n'était pas mutagène lors du test de mutation bactérienne inverse in vitro (test d'Ames) mais a induit une légère augmentation des aberrations numériques dans le test cytogénétique in vitro utilisant
des cellules de poumon de hamster chinois (CHL) avec activation
métabolique et des micronoyaux dans le test du micronoyau sur la moelle
osseuse de rat. Le mécanisme d'induction des micronoyaux était la
ségrégation de chromosomes anormaux (aneugénicité) et non un effet
clastogène sur les chromosomes.
Insuffisance de la fertilité
Aucune étude de fertilité chez les animaux n'a été réalisée pour
évaluer l'effet d'alectinib. Aucun effet indésirable sur les organes
reproducteurs mâle et femelle n'a été observé dans les études
toxicologiques en général. Ces études ont été conduites chez les rats
et les singes, à une exposition égale ou supérieure à, respectivement,
2,6 et 0,5 fois l'exposition chez l'Homme, mesurée à partir de l'Aire
Sous la Courbe (ASC), à la posologie recommandée de 600 mg deux fois
par jour.
Tératogénicité
Alectinib a induit des toxicités embryo-fœtales chez des rates et
lapines gravides. Chez les rates gravides ; alectinib a induit une
perte embryo-fœtale totale (avortement spontané) à des expositions
correspondantes à 4,5 fois l'exposition de l'ASC humaine ; des petits
fœtus au retard d'ossification et des anomalies mineures des organes à
des expositions correspondantes à 2,7 fois l'exposition de l'ASC
humaine. Chez des lapines gravides, alectinib a induit une perte
embryo-fœtale, des petits fœtus et a augmenté l'incidence de variations
squelettiques à des expositions correspondantes à 2,9 fois l'exposition
de l'ASC humaine à la dose recommandée.
Autres
Alectinib absorbe les ultra-violets (UV) entre 200 et 400 nm et a démontré un potentiel phototoxique dans un test in vitro de phototoxicité sur une culture de fibroblastes murins après une irradiation par UVA.
Dans les études toxicologiques à doses répétées, les organes cibles chez les rats et les singes lors d'une exposition cliniquement significative ont inclus, sans être limités à ces systèmes : le système érythroïde, le tractus gastro-intestinal et le système hépatobiliaire.
Une morphologie anormale des érythrocytes a été observée à exposition égale ou supérieure à 10- 60 % de l'exposition chez l'homme mesurée à partir de l'ASC à la posologie recommandée. L'extension de la zone de prolifération dans la muqueuse gastro-intestinale (GI) chez les deux espèces a été observée à exposition égale ou supérieure à 20-120 % de l'exposition chez l'homme mesurée à partir de l'ASC à la posologie recommandée chez les deux espèces. Une augmentation de la phosphatase alcaline hépatique (PAL) et de la bilirubine directe ainsi que de la vacuolisation/dégénérescence / nécrose des épithéliums des canaux biliaires et l'élargissement / nécrose focale des hépatocytes ont été observées chez les rats et / ou singes à exposition égale ou supérieure à 20-30 % de l'exposition chez l'homme mesurée à partir de l'ASC à la posologie recommandée.
Un effet hypotenseur léger a été observé chez les singes à expositions cliniquement significatives.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament
soumis à prescription hospitalière. Prescription réservée aux spécialistes en
oncologie ou aux médecins compétents en cancérologie. Médicament nécessitant une
surveillance particulière pendant le traitement.
Gélule.
Gélule blanche de 19,2 mm de longueur, portant la mention « ALE » imprimée sur la coiffe à l'encre noire et la mention « 150 mg » imprimée sur le corps à l'encre noire.
Plaquettes thermoformées (Polyamide/Alu/PVC/Alu) en aluminium/aluminium contenant 8 gélules. Taille de conditionnement : 224 (4 boîtes de 56) gélules.
Chaque gélule contient du chlorhydrate d'alectinib équivalent à 150 mg d'alectinib.
Excipient(s) à effet notoire :
Chaque gélule contient 33,7 mg de lactose (monohydraté) et 6 mg de sodium (laurylsulfate de sodium).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Lactose monohydraté
Hydroxypropylcellulose
Laurilsulfate de sodium
Stéarate de magnesium
Carmellose calcique
Enveloppe de la gélule
Hypromellose
Carraghénanes
Chlorure de potassium
Dioxyde de titane (E171)
Amidon de maïs
Cire de carnauba
Encre d'impression
Oxyde de fer rouge (E172)
Oxyde de fer jaune (E172)
Laque d'aluminium carmin d'indigo (E132)
Cire de carnauba
Gomme laquée blanche
Monooléate de glyceryle
"Vous souhaitez plus de pr�cisions sur Alecensa, Sophie Rozen et Val�rie Marsy sont � votre disposition pour vous accompagner sur le bon usage d'Alecensa et partager avec vous les documents sp�cifiquement �labor�s pour vous et vos patients".
valerie.marsy.vm1@roche.com�(0674407268) et�sophie.rozen@roche.com�(0607593070)
�